

12 days of Negative Results 🎄⛔🧪
Failures are still real science! 😜

We tried an experiment beyond the lab over the holidays: posting a 12-day Negative Results Advent Calendar on social media. By “negative results,” we mean both technical faceplants 🤦 (a protocol that breaks in an unexpected way) and genuine scientific dead ends ⛔ (a hypothesis that didn’t hold up). Both contain valuable information, but wouldn’t normally be included in a peer-reviewed paper.
Now we’ve collated the full calendar here, along with several honorable mentions that didn’t quite make the list the first time. We had great conversations and commiseration the first time, with people sharing stories of similar failures and offering helpful suggestions, and we’d love to hear more reactions! Diving into the nitty-gritty like this was genuinely fun, and we’d love to do (and see) more of this kind of open, behind-the-scenes science in the future.

The Calendar 🗓️⛔
⛔ result 1: We’re building an assay to measure perchlorate ☠️ in our Mars media so we can evolve microbes to clean it up 🧫 ❤️🩹 But the assay uses dichloromethane to extract the dye… which dissolves the bottom of polystyrene plates 😅🕳️ Switching to polypropylene fixed the issue. 🧪✅

This is us adapting a published assay to expand the dynamic range of a colorimetric perchlorate assay. Somebody suggested that polyurethane would work just as well as polypropylene - something for us to keep in mind! We also had some problems transferring dichloromethane with a multichannel pipette - it tends to drip.
⛔ result 2: We want to feed our Mars microbes acetate as a carbon source, since you can make it from CO₂ + water⚡️🌖 BUT when bugs metabolize acetate, the pH goes up instead of down like with sugar.🧪We buffered in the wrong direction and watched pH shoot from 8 → 10😅

This one is exciting. If acetate consumption increases pH, could you feed acetic acid to a bioreactor to keep the pH neutral and supply additional nutrients for fed-batch or continuous growth?
⛔ result 3: We used high salt as our first stressor to shake down our library selection & data collection pipeline🧂🧫But pellets from high-salt cultures are either rocks or snot globs. Our robot couldn’t resuspend them even after 300 pipetting steps with wide-bore tips🤖💪🚫

Someone in the comments mentioned this is probably due to excess EPS production and that we might be able to fix it by washing in PBS a few times first. Something to try!
⛔ result 4: Different LB mixes are NOT the same😅For our first year, we used HiMedia, then had to switch to Difco, which turned cloudy when we added salt and autoclaved🌫️🧂Now we’re on Fisher LB, which stays clear. But the level of salt inhibition is different in every media😒🧫
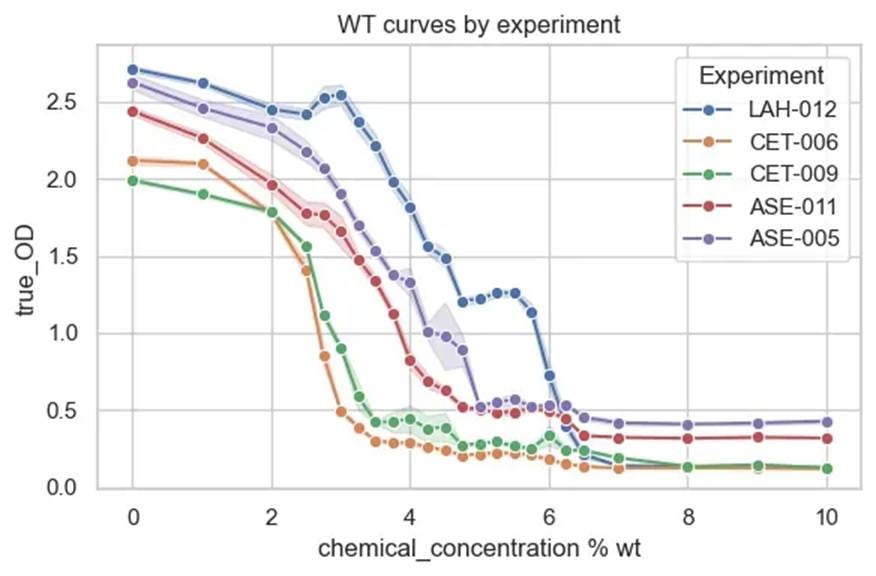
The X-axis here is % w/v salt added to LB-miller, which already has 1% salt in it, and the Y-axis is a 24-hour endpoint OD. All lines represent the same strain across five different days using different media. They vary in salt tolerance by almost 2%!
⛔ result 5: Because of our LB drama, we switched to measuring salt tolerance in M9, a defined minimal medium🧫➡️📏 And then…😅Strains that looked great in high-salt LB showed no improved salt tolerance in M9. Turns out “salt-tolerant” depends a lot on the exact media🧂🤷♂️
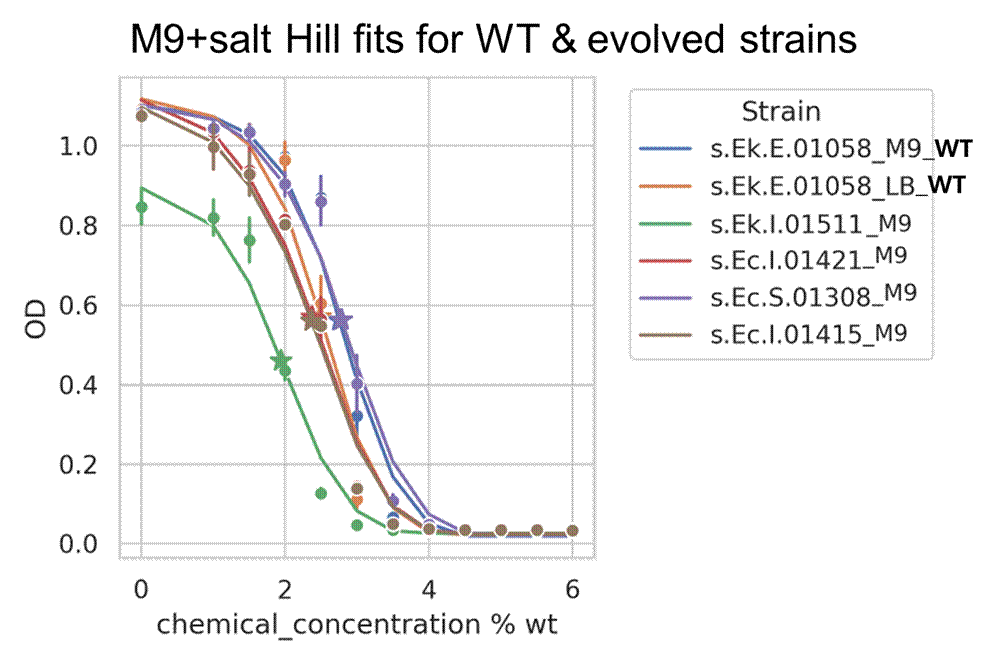
Someone shared their experience evolving a different, fast-growing E. coli strain in a turbidostat and sent us growth curves in LB + 12% salt for comparison with our results! Handy to help us understand what a truly evolved strain looks like.
⛔ result 6: Our Mars Media is meant to mimic mixing 40 g of regolith into 1 L of water, shake + filter 🚀🪨But when we try to make a concentrated stock, the calcium sulfate (gypsum) crashes out… and might be dragging the phosphate with it 😵💫🧪 #marsmilk

We don’t like to filter this media because then we don’t know the exact composition. To make higher mixes and represent putting less water into more regolith we’ve made altered recipes with reduced calcium & sulfate content. Media formulation is complicated!
⛔ result 7: Turns out trace metals really matter in minimal media 🧫🧪Once we started washing our starter cultures to remove carryover nutrients, they just… stopped growing unless we added trace metals 🤦♀️🧬 Super important detail for adaptive laboratory evolution! ⚙️🧬
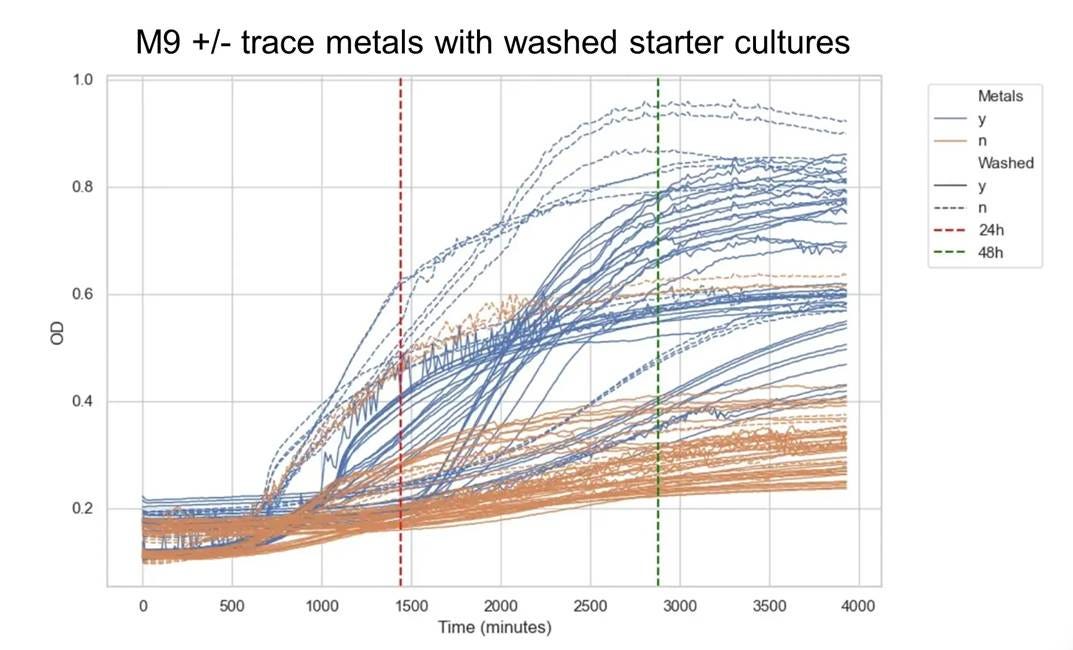
Just a few microliters of LB contain enough trace metals to support several subsequent cultures, so we didn’t see this problem until we started doing serial passaging in minimal media. Then our cultures slowly died off, and came right back when we supplemented with a trace metals mix. Now we’re building a regolith trace metal mix to represent what we think the real metal composition of sMRM is.
⛔ result 8: We found a mammalian expression plasmid (CMV promoter and all) as a contaminant in our bacterial selection experiments 😅🧬 Best guess? It hitchhiked in from the shared incubator-space centrifuge where folks do big DNA preps for mammalian cell culture 🚩🧪🙃
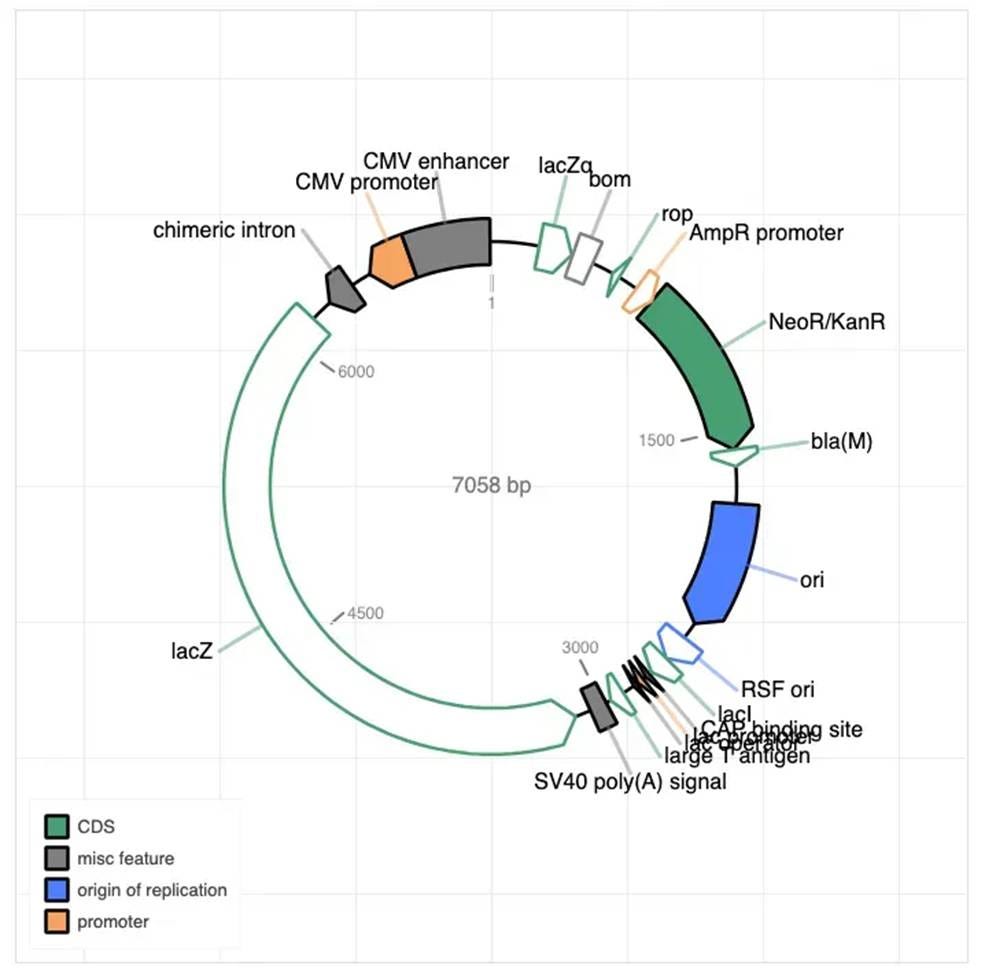
This plasmid didn’t have the priming regions to show up on our Illumina sequencing, but it kept showing up when we did nanopore sequencing - in libraries and elsewhere! We don’t use anything like it, so at first we thought it was contamination from our sequencing provider. They disagreed, and we tracked it down to a real contaminant that was all over the common lab spaces in the incubator.
⛔ result 9: We cloned libraries of tagmented gDNA into a suicide plasmid for transposition… only to discover carryover pUC plasmid in our commercial tagmentase 🧬😅 Leftover from protein purification, it totally dodged the suicide selection and snuck into our experiment 🕵️♂️🧪🛠️
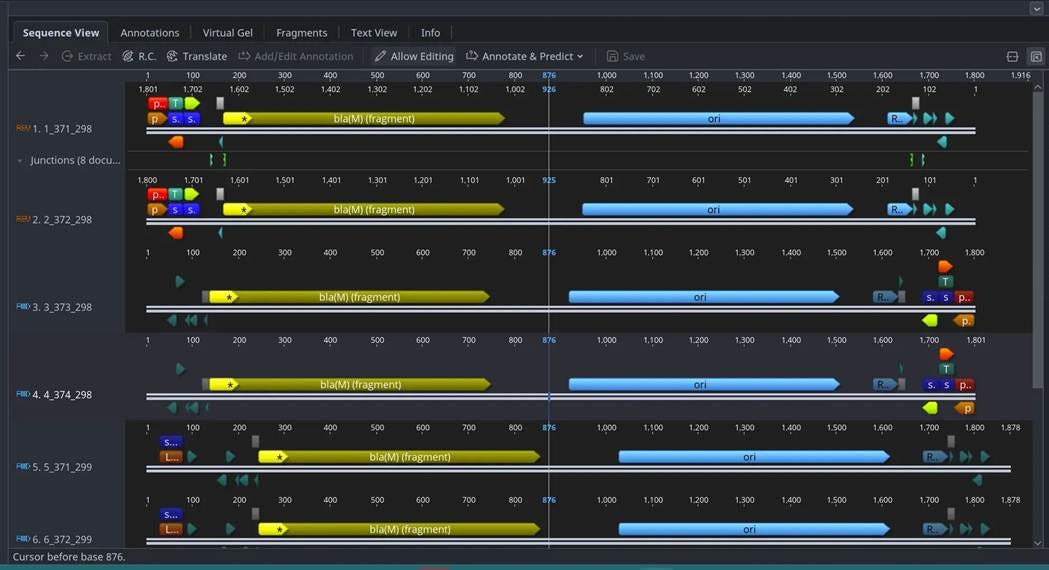
The big consequence here is that we can’t electroporate these libraries into our final host strain, since plasmids with replicative origins get a massive boost compared to plasmids that need to integrate into the genome during electroporation. This bias doesn’t seem to occur with conjugation, so it doesn’t ruin these libraries; it just means the final integration of the library genome needs to happen via conjugation, not electroporation. Conjugation works with more species, so we were probably going to do this anyway!
⛔result 10: For genomic integration in C. necator, we do repeated rounds of integration & excision🧬🔁That means days of growth without selection, and C. necator is so slow-growing it usually gets contaminated 😭 Other strains outgrow hitchhikers, but not this little guy🐌🧫
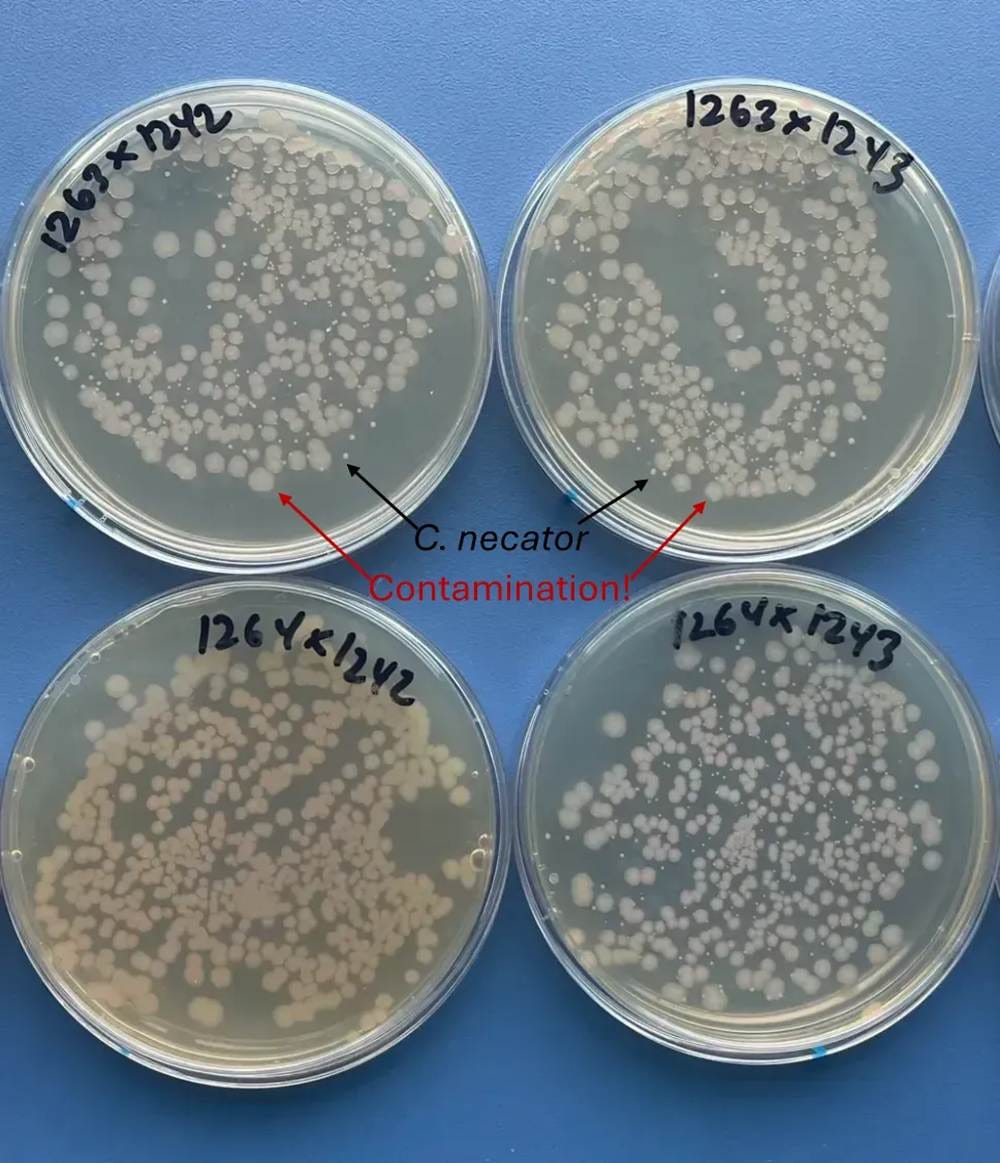
Standard bacterial sterile technique isn’t enough here! This protocol involves a week of growth and several passage steps in rich media without antibiotics, and contamination at any point ruins the whole process. You need to follow sterility protocols that are similar to those required for mammalian stem cell culture to entirely avoid contamination.
⛔ result 11: We barcode the genome and use Illumina reads to track fitness of 5e6 strains at once 🧬📊 But at that scale you need to add µg’s of gDNA to avoid bottlenecks. We didn’t, so our variance ended up far above Poisson noise 🤦♀️📉🎲
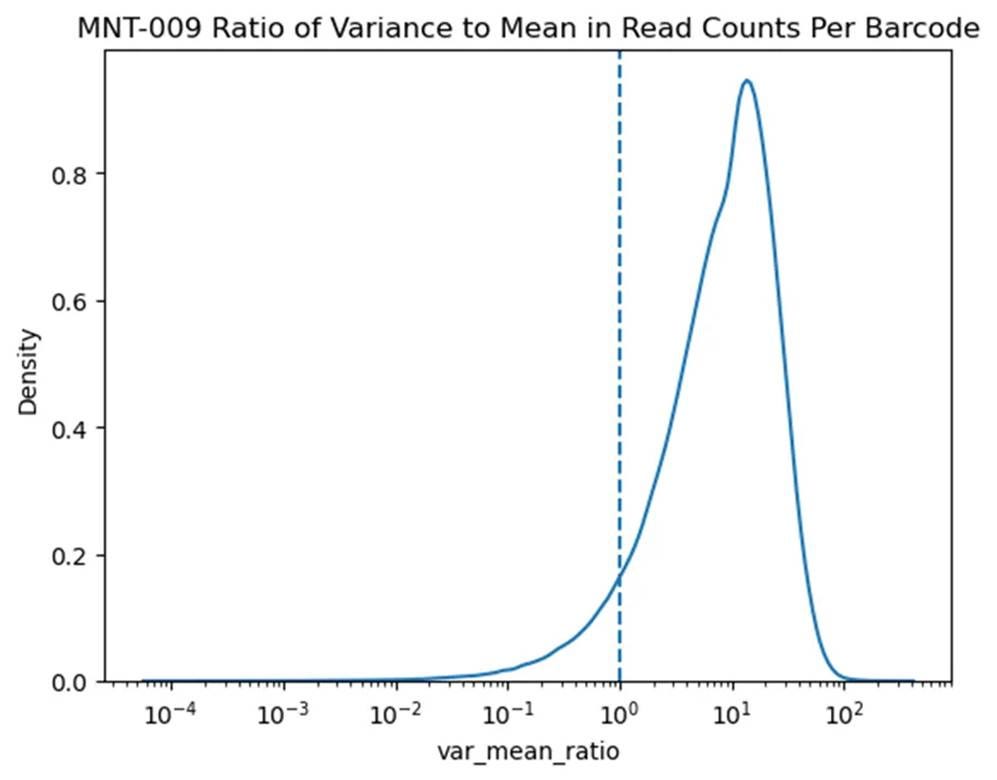
This is a wonderful example of a new QC metric exposing a specific problem. If we’re doing random sampling from an underlying distribution, then the variance should be equal to the man. If the variance is higher, it means we’re bottlenecking the underlying distribution somewhere! Once we knew this we could go through every step to figure out where the issue was - in this case during our primarily illumina PCR step. When you barcode genomes, you have a lot of irrelevant DNA. To get 50x barcode coverage you need about a nanogram of plasmid DNA, but when your barcode genomes that requires more than a microgram!
⛔ result 12: Some of our libraries are already pretty skewed before we start selection 😅📊 Not always bad enough to trash, but it means we need much bigger bottlenecks to avoid chopping off all the low-abundance members 🧬🔍🧵
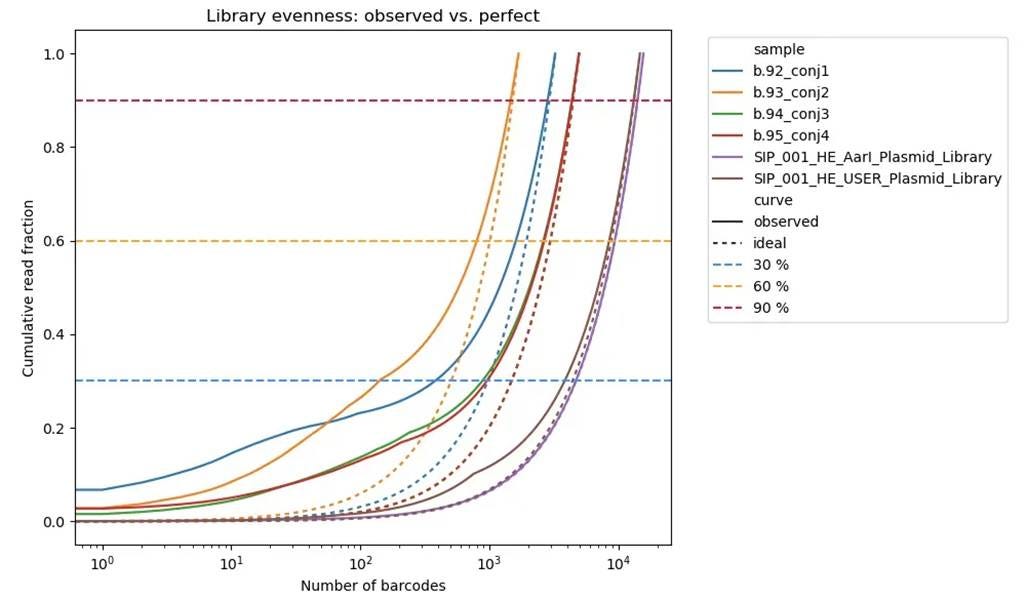
These libraries go through a lot of processing steps before selection, and it’s crucial to minimize any skew from those steps. Selection is supposed to create skew that carries information we can measure, but if the library is already skewed at the start, we lose dynamic range and have lower sensitivity to real fitness effects.
Honorable Mentions🥈🧫
In brainstorming this calendar, we had nearly 30 ideas, but not all of them made sense without a lot of context. Here are a few of the runner-ups we ended up cutting out.
⛔ result X: Tris is a bad buffer for cell culture 😬 We tried it in our Mars media to avoid phosphate buffer (it was already on the shelf), but Tris can cross membranes at high pH and mess with DNA & enzymes 🧬🧪 After a few experiments, we ditched it and switched to TES ✅
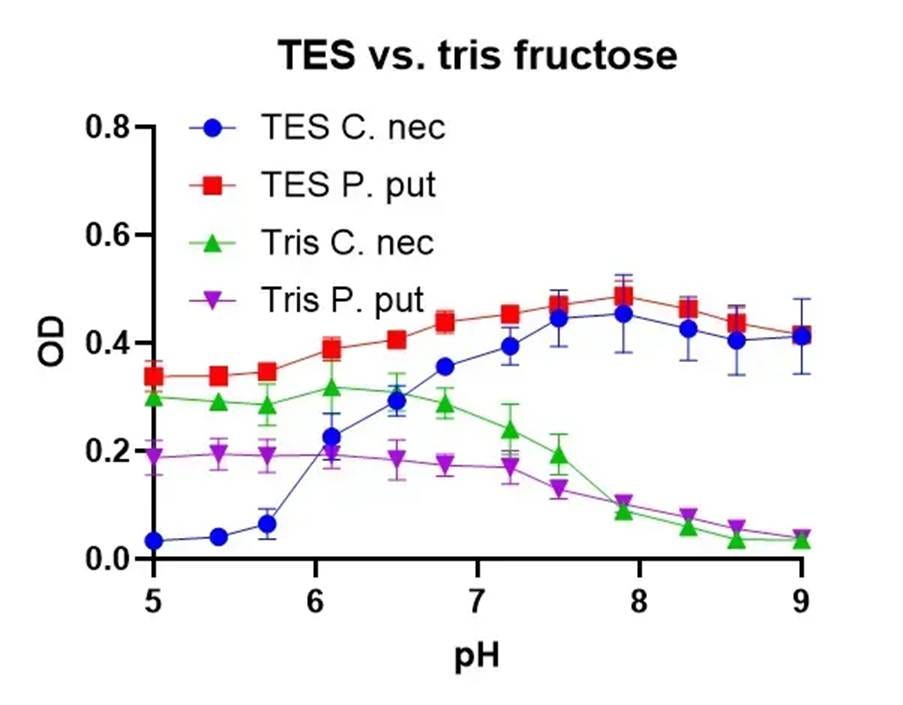
This was a simple lesson to learn. Don’t use Tris in cell culture! Use one of the Good Buffers instead. We’re not entirely sure why C. necator didn’t grow well below pH 7 in TES here. 🤷
⛔ result X+1: We sent our lab DI water to an external lab for ion analysis 🧪📦 and it came back with 2.5 ppm iron 😳🧲That’s ~2.3 µS conductance, still in the “DI” purity range ✅💧 but way more Fe than we expected. Remember, DI water isn’t truly pure 🤦♀️✨
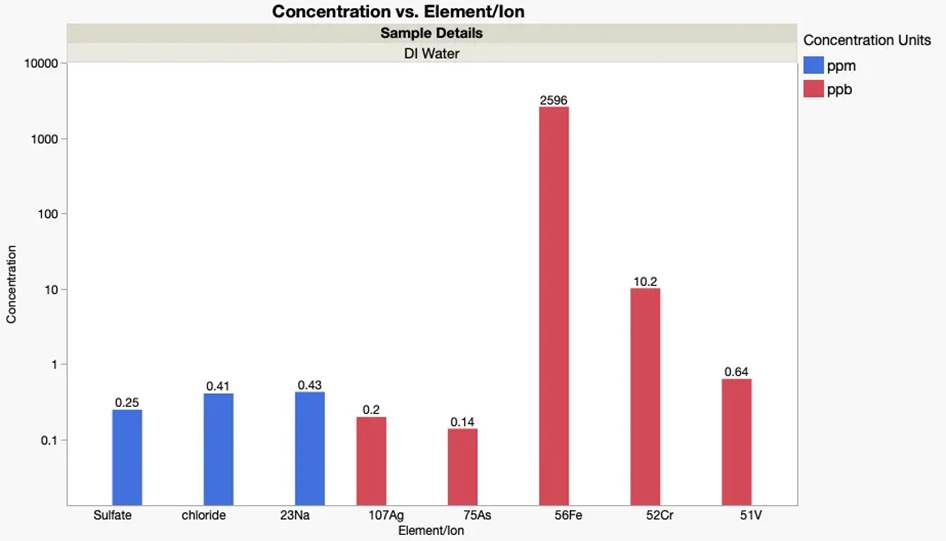
Normally “DI” water is 5-1 µS, “MilliQ” water is 1-0.1 µS, and “ultrapure” water is below 0.1 µS. How pure does your water need to be? We re-measured this in our new space, and it’s much lower, with no detectible iron and a total conductivity around 0.12 µS. ✅
⛔ result X+2: We use DAP auxotrophy to kill off the donor strain after library conjugation 🧬🧫🚫…but our full-scale libraries included fragments with dapA, so donor cells grabbed them and survived 😅🦠✅Congrats to us: we accidentally ran an auxotrophy complementation experiment 🤦♀️🧪
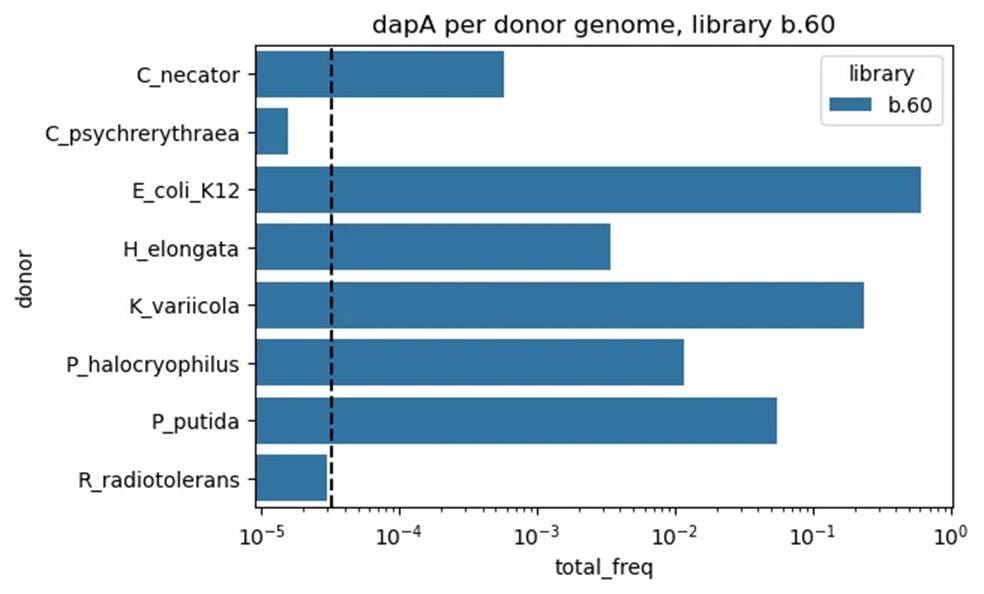
We turned this accident into its own experiment in our previous post, if you’re curious about more details.
⛔ result X+3: To stop DAP-auxotrophy escape, we made our donor strain double auxotrophic—it needs DAP + 5-ALA to survive 🧬🧫🔒 But 5-ALA has to be really well washed out… and if it isn’t, donor cells carrying dapA fragments can sneak through for several passages 😅🦠➡️🧪
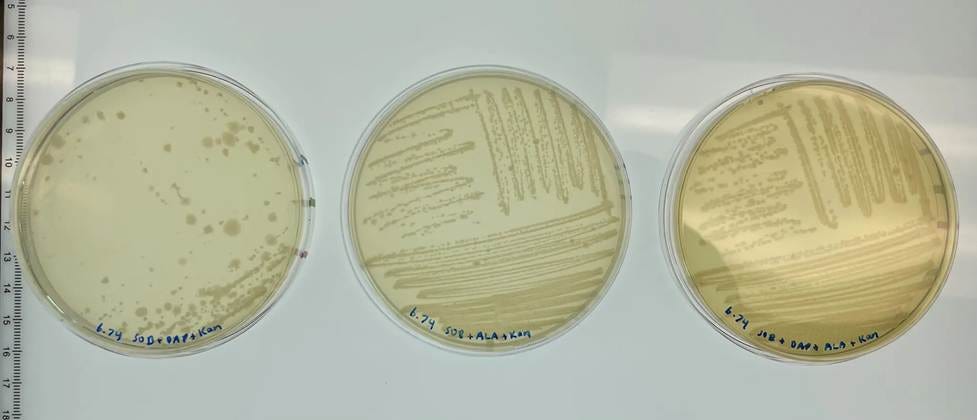
Here, we restreaked our freshly conjugated libraries onto +DAP, +ALA & +ALA & DAP. There should be a lot of growth on the plates with both supplements, but the plates with only a single one should have fairly minimal growth. But excess growth on the +Ala plate tells us that the conjugation carried over a lot of cells that require Ala to survive. 🤔 Washing the conjugation a couple of extra times fixed the issue.
⛔ result X+4: We do tons of high-efficiency electroporations on a GenePulser Xcell ⚡️🧫But if the shockpod lid isn’t fully closed… it will pretend it zapped your cells 😅 It prints a time constant and everything ⏱️✅ but delivers no shock 🙃Mystery “why won’t this transform?” solved 🤦♀️🔬

The electroporator is now covered with warnings to close the lid before you use it. ⚠️
⛔ result X+5: Turns out going from a strain name ➡️ a genome accession ID is way trickier than it sounds 😅🧬And if you want to automate it for hundreds/thousands of genomes? It becomes a whole pipeline problem 🤖📚🔍
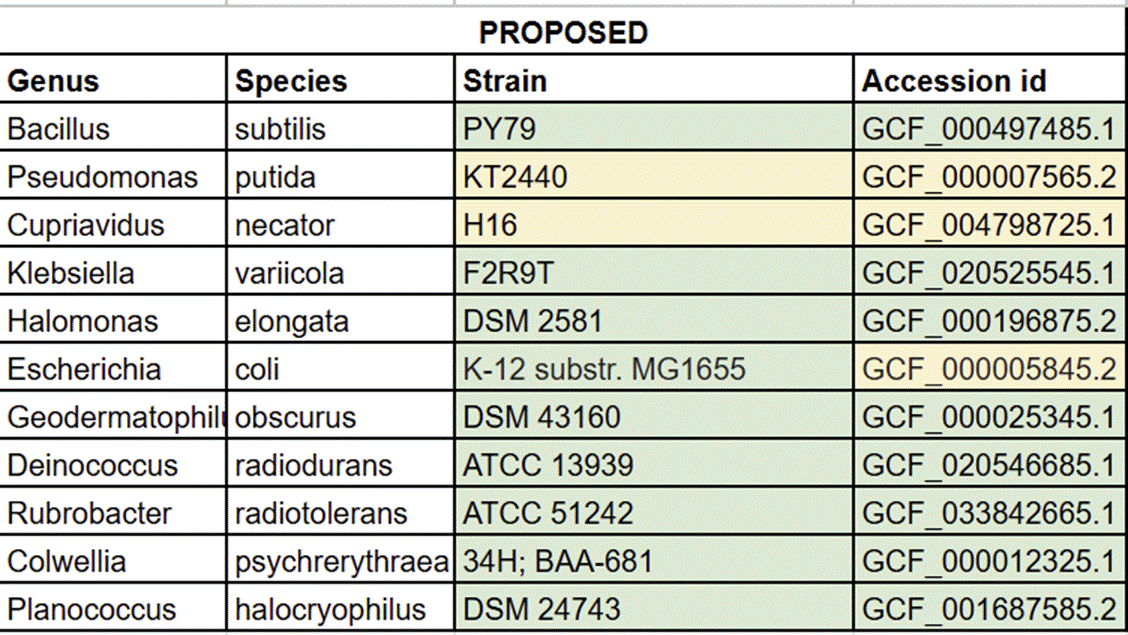
We have multiple different strains of P. putida, C. necator, and E. coli, so the yellow highlights are notes to check those fields against the strains we actually use! ✅
Closing Thoughts 🧠✨
This 12-day Negative Results Advent Calendar was a cathartic exercise for us and helped us reflect on the past year of experiments. Science isn’t a clean, linear path to discovery; it’s a messy, iterative process often riddled with tiny, often hilarious, technical failures.
Inviting people behind the curtain and getting a positive response showed us there’s a real hunger for this kind of open, behind-the-scenes scientific communication. Being transparent about failure can be just as informative, and definitely more fun, than only celebrating success!
Comment with your thoughts on our negative results, or share some of your own!